
Molecular electronics
In the recently published article titled Combining Multiscale MD Simulations and Machine Learning Methods to Study Electronic Transport in Molecular Junctions at Finite Temperatures (J. Phys. Chem. C 2021, 125, 36), Rafał Topolnicki, Robert Kucharczyk, and Wojciech Kamiński present results of their studies on the application of neuron networks in modelling of the influence of temperature on transport properties of organic molecular junctions.
Molecular electronics is an attractive alternative to currently used traditional semiconductor electronics devices, enabling in particular further progress in their miniaturisation. Still, however, an array of phenomena related to electronic transport through organic semiconductor elements requires them to be better known from the theoretical side.
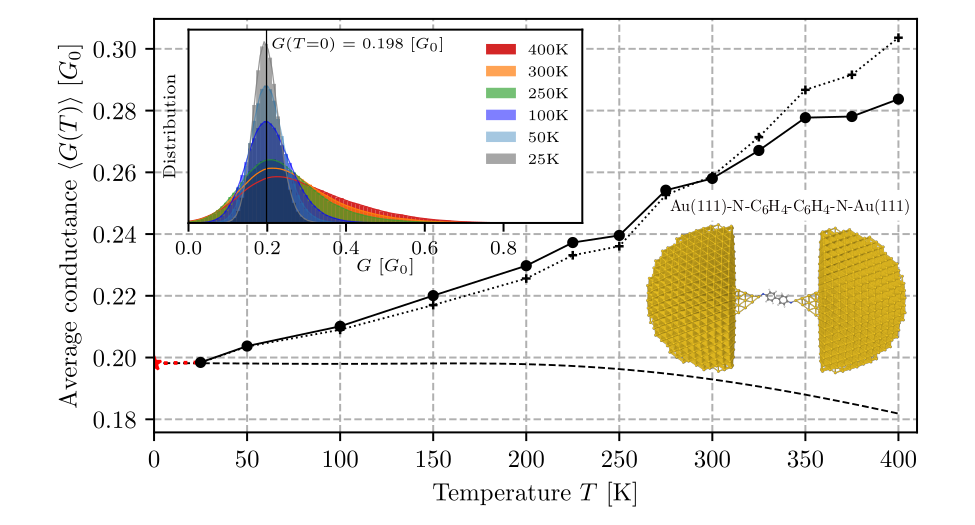
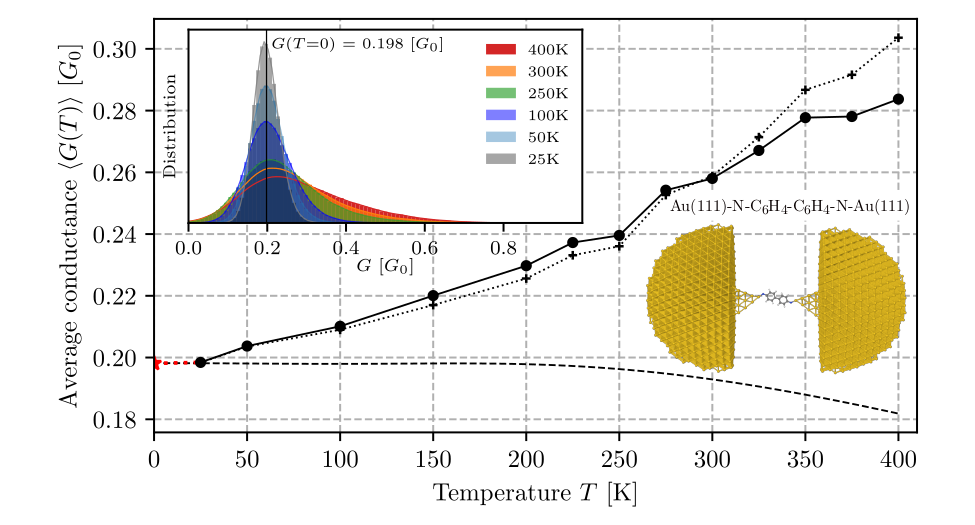
Experimental studies on transport properties of molecular systems are most commonly done at room temperature, and during a single measurement the geometry of a junction evolves as a result of thermal fluctuations of a molecule, during which the structural and electronic parameters of a junction change in an uncontrollable way. Change of each of those parameters affects the conductivity. The result of a single experimental measurement is therefore averaged after many various conformations of an active molecular element. Theoretical studies are usually based on calculations of transmission spectra for a fixed junction geometry corresponding to the basic state of the system at absolute zero temperature. Thus, such methodology does not allow to take into account temperature-induced changes in the transport properties of the junction. The mentioned discrepancies between experimental and model conditions contribute to significant differences between the results of calculation and experiment.
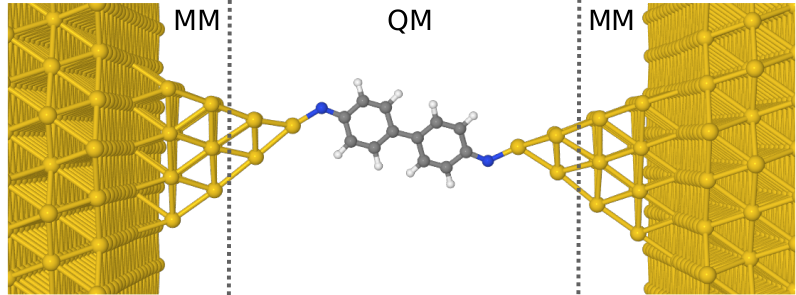
The paper proposes a method of making the theoretical description realistic through development and implementation of new accounting solutions that pin down three complementary computing techniques. Using the QM/MM molecular dynamics method, long-term simulations of the structural evolution of Au(111)-S-C6H4-C6H4-S-Au(111) and Au(111)-N-C6H4-C6H4-N-Au(111) junctions were carried out at temperatures from 25 K to 400 K, then using active machine learning methods, a method was proposed to select representative atomic configurations for which time-consuming electron transport calculations are performed (using the nonequilibrium Green’s function method, NEGF). The resulting data were used to teach a neural network to best predict the tunnel current based on the structural and electron characteristics of the junction. Next, tunnelling current predictions were made for molecular dynamics trajectories at different temperatures. In this way, the temperature dependence of the tunnelling current and the contribution to this dependence due to thermal changes in the conformation of the organic molecule were determined.
Added by: Joanna Molenda-Żakowicz
Dean’s representative for faculty promotion and media relations


{kind=link}